Joint Research Group Macromolecular Crystallography
Structure of the month - October 2014
Nature Communications 5 (2014) 4626.
ATP-induced electron transfer by redox-selective partner recognition
Sandra E. Henniga, Sebastian Goetzla, Jae-Hun Jeounga, Martin Bommera, Friedhelm Lendzianb, Peter Hildebrandtb, Holger Dobbeka*
a Humboldt Universität zu Berlin, Institut für Biologie, Strukturbiologie/Biochemie, Unter den Linden 6, 10099 Berlin, Germany
b Technische Universität Berlin, Institut für Chemie, Sekr. PC14, Strasse des 17. Juni 135, 10623 Berlin, Germany
* Corresponding author: Holger Dobbek, Humboldt Universität zu Berlin, Institut für Biologie, Strukturbiologie/Biochemie, Unter den Linden 6, 10099 Berlin, Germany
E-mail: holger.dobbek@biologie.hu-berlin.de
Phone: +49(0)30-2093-6369
Abstract
Enzymes provide a scaffold of chemical groups in precise arrangements, which allow them to lower the activation energy of chemical reactions. Such molecular machines are able to carry out highly energy-demanding chemical processes at atmospheric (or physiological) conditions. More frequently than desired an enzyme is inactivated by an unwanted side reaction. In living organisms, where the synthesis of the enzyme and cofactor also presents a sizable up-front investment, repair systems exist for crucial enzymes.
A globally important process is the assimilation of atmospheric CO2 into organic matter. Under anaerobic conditions, CO2 may be assimilated to acetate through the microbial Wood-Ljungdahl pathway. "The corrinoid (B12) and [4Fe-4S] cluster containing protein (CoFeSP) acts in the late steps of the pathway. CoFeSP accepts a methyl group from a methyl-tetrahydrofolate bound methyltransferase and transfers it to acetyl-CoA synthase, for which its B12 cycles between the CH3-Co3+ and Co1+ active states. The energy-rich Co1+ state of CoFeSP is prone to oxidation generating the [inactive] Co2+ state." Hennig & Jeoung et al. have previously identified an ATP-dependent rescue factor RACo (reductive activator of CoFeSP), which is able to return the B12 cofactor to the active Co1+ state (Structure of the Month November 2012, PNAS 2012). The authors now present the structure of the RACo-CoFeSP complex (Fig. 1) and propose a unique model for the ATP-dependent electron transfer to B12, which is initially energetically uphill (Fig. 2). For this purpose, RACo manipulates the coordination state of the cofactor. Commonly, the active Co1+ is tetra-coordinated (the B12 corrin ring provides all four ligands), while inactive Co2+ is penta-coordinated with one external (e.g. water) ligand. RACo replaces the β-ligand with the side chain of its own serine 398, while encapsulating the B12 and locking out water. In an ATP-energised conformational change the serine ligand is then forced from Co2+ in the absence of an alternative ligand, thus transiently creating an unstable tetra-coordinated Co2+, which is easily reduced to Co1+ by an electron from RACos [2Fe-2S] cluster. (M.B. & S.H.)
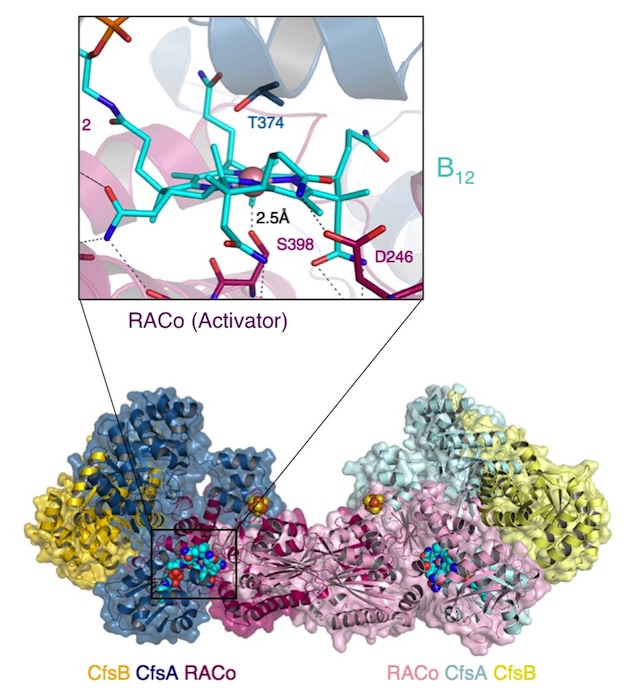
Fig. 1. Dimer of the CoFeSP (enzyme) - RACo (activator) complex. The CoFeSP enzyme is shown as blue (large subunit) and yellow (small subunit) containing the (vitamin-)B12 cofactor. A dimer of RACo (pink) docks tightly with CoFeSP and clamps its B12 cofactor, shielding it from solvent (inset). Crucially, the β-ligand of B12 is replaced by the hydroxyl group of Ser-398 from RACo.
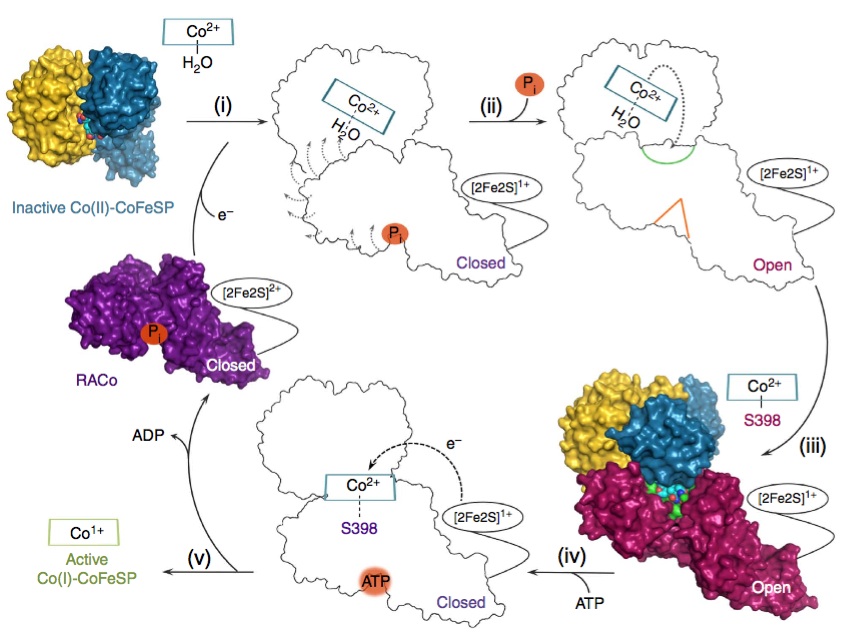
Figure 2. Proposed model for CoFeSP reactivation. Inactive CoFeSP-CO2+ is selectively bound by RACo (i), leading to the opening of its binding site for B12 (ii) and replacement of the β-ligand water with Ser-298 in RACo (iii). Ser-398 is then forced away from B12 through an ATP-driven movement in RACo (iv), transiently creating a tetra-coordinated Co2+ in B12, which is easily reduced to active Co1+ (v).
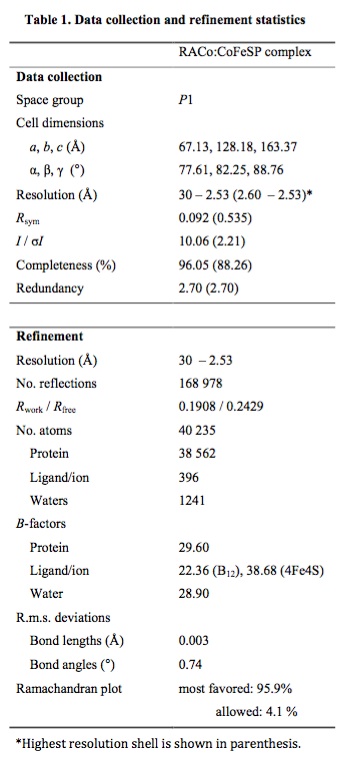
Table 1.